CASE REPORT | https://doi.org/10.5005/jp-journals-10067-0027 |
A Case of Tuberous Sclerosis Triad with Psychiatric Manifestation
1–3Department of Psychiatry, Silchar Medical College and Hospital, Silchar, Assam, India
Corresponding Author: Prosenjit Ghosh, Department of Psychiatry, Silchar Medical College and Hospital, Silchar, Assam, India, Phone: +91 9435072563, e-mail: p_ghosh72@yahoo.com
How to cite this article Ghosh P, Doley M, Gogoi A. A Case of Tuberous Sclerosis Triad with Psychiatric Manifestation. Ind J Priv Psychiatry 2019;13(1):18–21.
Source of support: Nil
Conflict of interest: None
ABSTRACT
Tuberous sclerosis (TS) is a rare disorder of genetic origin, caused by mutations in the genes TSC1 and TSC2. It is characterized by benign tumors with multisystem involvement resulting in dermatological, nephrological, neurological and psychiatric manifestations. We are presenting a case of a 21 years old male with tuberous sclerosis presenting with dermatological manifestations, seizures, intellectual disability and psychiatric manifestations including delusions and hallucinations, developing extrapyramidal symptoms after antipsychotic administration. Neuroimaging showed characteristic subependymal calcified nodules. With appropriate anticonvulsant and antipsychotic medications he showed clinical improvement.
Keywords: Extrapyramidal symptoms, Intellectual disability, Psychosis, Seizure, Subependymal nodules, Tuberous sclerosis, Vogt’s triad.
INTRODUCTION
Tuberous sclerosis (TS) complex is a genetically determined multisystem disorder that may affect any human organ system. It was initially described by Rayer in 1835 and then by von Recklinghausen in 1862.1 Bourneville coined the term tuberous sclerosis to describe the brain pathology. Two genes causing TSC have been identified: TSC1 (9q34) and TSC2 (16p13.3). Their protein products hamartin (TSC1) and tuberin (TSC2) form a complex that regulates the PI3-kinase/AKT pathway to control cell size and proliferation. Loss of these cellular functions is central to the pathogenesis of hamartomas associated with TSC.2 Its incidence is 1 in 6,000–10,000 live births.1
International Tuberous Sclerosis Complex Consensus Conference (2012) recommended genetic and clinical criteria for diagnosis of tuberous sclerosis. The genetic criterion consists of a pathogenic mutation in the TSC1 or TSC2 genes. The clinical criteria consist of 11 major and 6 minor (Table 1) criteria. Clinically, definite diagnosis: 2 major features or 1 major with ≥2 minor features, possible diagnosis: 1 major feature or ≥2 minor features.1
The classical Vogt’s triad consists of: epilepsy, low intelligence and adenoma sebaceum (epiloia) (Reddy et al.).4
Tuberous sclerosis complex is associated with significant psychiatric comorbidities like anxiety, depression, aggression, ADHD, disruptive behavior and are frequently reported.5 However, comorbidities like cases of psychosis, schizophrenia, bipolar disorder, auditory hallucinations, alcoholism are less frequently reported.6
Here, we are presenting a casereport of tuberous sclerosis complex with comorbid psychosis presenting with antipsychotics induced extrapyramidal symptoms (EPS).
CASE DESCRIPTION
A 21-year-old male from rural background presented to Psychiatry Department of Silchar Medical College and Hospital on 15-04-2019 during emergency hours with a history of insidious onset of abnormal behaviors for 5 months, which were exacerbated in the preceding couple of weeks, without any major life-event or stress preceding these symptoms. Abnormal behaviors reported included fearfulness, muttering, smiling to self, anger outbursts without any apparent reason, hearing voices not heard by others and self-injurious behavior in the form of beating himself. All these symptoms occurred in the backdrop of a history of episodic jerky movements of limbs with tonic contractions and associated loss of consciousness.
| Major features | Minor features |
|---|---|
| • Hypomelanotic macules (≥3, at least 5 mm diameter) | • “Confetti” skin lesions |
| • Angiofibromas (≥3) or fibrous cephalic plaque | • Dental enamel pits (>3) |
| • Ungual fibromas (≥2) | • Intraoral fibromas (≥2) |
| • Shagreen patch | • Retinal achromic patch |
| • Multiple retinal hamartomas | • Multiple renal cysts |
| • Cortical dysplasias* | • Nonrenal hamartomas |
| • Subependymal nodules | |
| • Subependymal giant cell astrocytoma | |
| • Cardiac rhabdomyoma | |
| • Lymphangioleiomyomatosis (LAM)† | |
| • Angiomyolipomas (≥2)† |
* Includes tubers and cerebral white matter radial migration lines
† A combination of the two major clinical features (LAM and angiomyolipomas) without other features does not meet criteria for a definite diagnosis
On detailed enquiry, informants gave history, supported by documents, that he had been admitted in SMCH Medicine Department in Silchar Medical College Hospital on 09-01-2012 for appearance of nodules in face, hypopigmented lesion in whole body with episodes of involuntary jerky movements of the limbs, lasting a few seconds along with urinary incontinence at times and brief staring spells. He was then diagnosed as a case of tuberous sclerosis and was put on tab phenytoin 300 mg per day in divided dose and oral multivitamin and was discharged on 16-01-2012 with same medications. He was completely seizure free for subsequent 5 years with daily oral phenytoin of 300 mg.
However, starting from 2017, he started having occasional breakthrough seizures while on the previous medication. His latest seizure attack had been on 01-04-2019, i.e., two weeks prior to this presentation.
Mental state examination revealed inadequate eye contact, poor personal hygiene, blunted affect with delusion of persecution and auditory hallucinations. He was provisionally diagnosed as a case of other mental disorders due to brain damage and dysfunction and to physical disease (F06 according to ICD-10). Tab phenytoin was stopped, and he was initiated with tab sodium valproate 800 mg per day in divided doses, tab risperidone (3 mg) and trihexyphenidyl hydrochloride (2 mg) once daily and tab lorazepam 2 mg at bedtime.
After a few days patient was bought again to SMCH psychiatry OPD with complaints of agitation, restless feeling, increased anger, tremulousness of hands, decreased oral intake, increase salivation, slowness of daily activities, along with fearfulness and hearing of voices. These symptoms appeared after taking medication prescribed on 15-04-2019 for 5–7 days. So, he was admitted in male psychiatry ward for detailed evaluation.
Further evaluation following admission revealed that he was born of a nonconsanguineous marriage, was delivered vaginally at home following an uneventful labor with no postnatal complications. He showed delay in developmental milestones—walking without support at 2 years of age, speaking monosyllables at 3 years of age. He used to remain aloof and self-absorbed in his childhood. He was admitted in primary school at 6 years of age, but due to problem in coping up with peer groups as well as poor academic performance, he was dropped out from school. At 9 years of age, he started developing small nodules over his nose and gradually multiple nodules appeared all over his facial region along with multiple hypopigmented lesions on his body (3 in back, 2 in upper thighs). Along with this he had first episode of seizure in 11 years of age lasting 2–3 minutes. Clusters of attacks occurred at 2–3 weeks intervals. He developed brief staring spells, occurring 2–3 times per month. He walked independently, could follow simple commands, could feed himself, could perform daily routine activities of self-care without assistance. He spoke minimally but with history of hypergraphia. For a major part of the day he scribbled and drew scenic pictures and human figures when provided with pencils and paper. He did not have any formal training in painting. There was no family history of any neurological or psychiatric disorder. But his father had a few nodules on his face and nasal bridge in a scattered fashion.
Physical examination findings were like this: blood pressure = 110/70 mm Hg, pulse rate = 82/minute. There was pallor, but no icterus, cyanosis, nail abnormalities, or palpable neck or axillary lymph nodes. He had bilateral fine hand tremors, bradykinesia, angiofibromatous nodules over whole of oro-facial region including nose and forehead. Hypopigmented lesions (total 5 in number) were evident over his lower back and thighs. He also had multiple crinkled skin lesions (Shagreen patches) over his back.
Mental state examination revealed average built, poor hygiene, decreased psychomotor activity, with the patient being uncooperative, and rapport could not be established. Speech was slurred, was of low tone, and was irrelevant at times. Mood was fearful with moderately apprehensive, appropriate, reactive, labile affect with a wide range. Thought revealed delusion of persecution, which was encapsulated and well-systematized. Auditory hallucination was present. His intelligence was low (IQ = 60) with impaired abstract thinking, judgement and reasoning and insight being grade I (Figs 1 to 3).
Routine laboratory investigation findings were within normal range except low Hb (7.8 g/dL).
Among radiological findings, CT-scan brain showed multiple subependymal calcified nodules bilaterally in the region of body of lateral ventricles (Fig. 4). Chest X-ray, ECG and USG whole abdomen showed no abnormality. Ophthalmological findings were normal.
Provisionally the patient was diagnosed according to ICD-10 criteria as other mental disorders due to brain damage and dysfunction and to physical disease (TSC) with mild mental retardation (F06 with F70) with acute drug induced EPS.
He was started with IV infusion fluids, injectable multivitamin, trihexiphenidyl hydrochloride 4 mg orally per day in divided doses and sodium valproate 800 mg orally per day in divided doses. Patient’s restless agitated behavior decreased and gradually his hand tremors and bradykinesia along with slurring of speech decreased. He was then discharged with tab olanzapine 10 mg, tab sodium valproate 800 mg in divided doses and tab trihexyphenidyl hydrochloride 2 mg. On first follow-up visit after 10 days, his psychotics symptoms were found to be improved but not significantly. He was seizure free. However, he again developed EPS. He was then advised tab risperidone 2 mg and tab trihexyphenidyl 2 mg per day along with sodium valproate in the earlier dose. Later, after around 6 weeks he developed a breakthrough seizure episode when he was again brought to our OPD. By that time his psychotic symptoms showed improvement. His valproate dose was increased to 1,000 mg per day in divided doses. In subsequent visits, the risperidone dose was gradually reduced. Now he is maintaining well with risperidone 0.5 mg per day, valproate 1,000 mg per day, seizure free. Now he is more concerned with body dysmorphism, mainly facial. Patient and attendant were counselled regarding this.
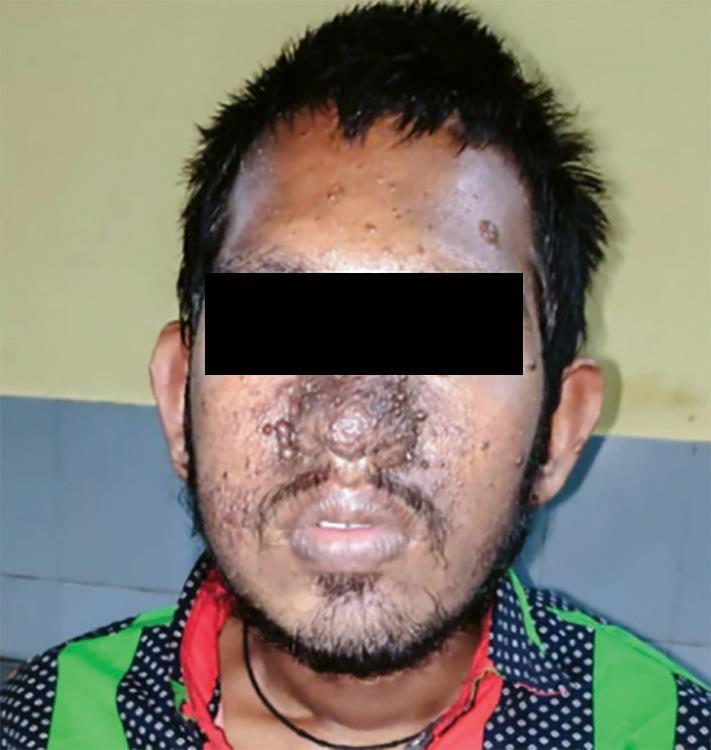
Fig. 1: Facial angiofibromas (adenoma sebaceum)
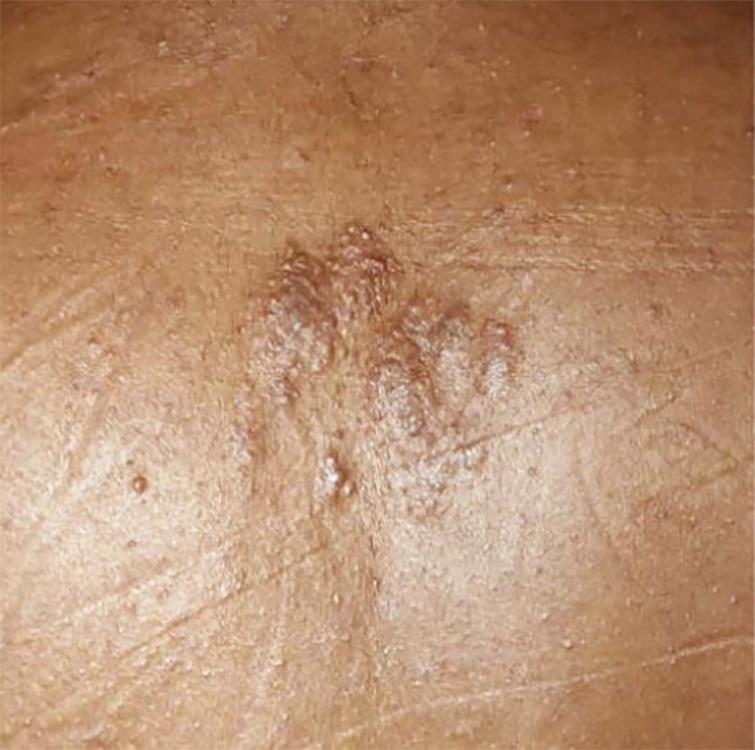
Fig. 2: Shagreen patch over back

Fig. 3: Hypomelanotic macule over lower back

Fig. 4: CT brain: Calcified subependymal nodules
DISCUSSION
Early diagnosis of TS is important through clinical, radiological evaluation. Regular monitoring of the individual is very important, as it affects all the system of the body. In this case, development of symptoms is in sequences as follows—skin lesion at 9 years of age, first seizure episodes at 11 years of age followed by cognitive decline, changes in temperament, behavior and finally psychotic symptoms. Psychotic symptoms are not uncommon in children with tuberous sclerosis; a study on 90 children reported more than 50% having psychotic behavior.7 About 62% of patients with TS have some form of epilepsy.8 In a range of 38% (Webb)9—64% (Gillberg)5 presented with intellectual disability and cognitive impairment. A study by Ebrahimi-Fakhari et al.,10 the majority of patients presented with CNS involvement—cortical dysplasia in 51.5%, subependymal nodules in 47.7% and seizure disorder overall in 69.8%.11 Dermatological manifestations like angiofibroma, shagreen patches are recognized manifestation in adults10 as in this case report.
TS affects both patient and family physically and psychologically. It is considered a rare disorder. There is no specific treatment for the disease but a 20th month follow-up study of 113 patients reported that antipsychotic medication can improve the mental symptoms and stabilize the mood of individuals with TS (Chung et al.).3 Current guidelines on the management of epilepsy in TS emphasizes greatly on early recognition and prompt treatment of epilepsy.12 Multiple drug therapies are ongoing and research projects are being carried out in the world for further workup of the genes involved and treatment strategies.
CONCLUSION
Psychological intervention and pharmacotherapy combined brought seizure and psychotic process under control in this case. Patient was on regular follow-up. Patient reported feeling of anxiousness, body dysmorphism. Reassurance was given. As the patient was hypergraphic, he was encouraged to culture his skills in a better way. As he is having mild intellectual disability, cognitive enhancement training is advised. Parents were made to understand about the disease and be aware of the emotional needs of the child. Further, they were advised to continue the medication and warning signs of seizure and abnormal behavior was explained. Necessity of regular follow-up visit was also explained.
REFERENCES
1. Northrup H, Krueger DA, Roberds S, et al. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol 2013;49(4):243–254. DOI: 10.1016/j.pediatrneurol.2013.08.001.
2. Kwiatkowski DJ, Whittemore VH, Thiele EA, ed. Tuberous sclerosis complex: genes, clinical features and therapeutics. John Wiley and Sons; 2011.
3. Chung TK, Lynch ER, Fiser CJ, et al. Psychiatric comorbidity and treatment response in patients with tuberous sclerosis complex. Ann Clin Psychiatry 2011;23(4):263–269.
4. Reddy NS, Chandrakala S, Prasad BN, et al. A rare case report on tuberous sclerosis. Pharma Tutor 2015;3(12):29–31.
5. Gillberg JC, Gillberg C, Ahlsén G. Autistic behaviour and attention deficits in tuberous sclerosis: a population-based study. Dev Med Child Neurol 1994;36(1):50–56. DOI: 10.1111/j.1469-8749.1994.tb11765.x.
6. Muzykewicz DA, Newberry P, Danforth N, et al. Psychiatric comorbid conditions in a clinic population of 241 patients with tuberous sclerosis complex. Epilepsy Behav 2007;11(4):506–513. DOI: 10.1016/j.yebeh.2007.07.010.
7. Hunt A, Dennis J. Psychiatric disorder among children with tuberous sclerosis. Dev Med Child Neurol 1987;29(2):190–198. DOI: 10.1111/j.1469-8749.1987.tb02135.x.
8. Webb DW, Fryer AE, Osborne JP. On the incidence of fits and mental retardation in tuberous sclerosis. J Med Genet 1991;28(6):395–397. DOI: 10.1136/jmg.28.6.395.
9. Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci 1991;615(1):125–127. DOI: 10.1111/j.1749-6632.1991.tb37754.x.
10. Ebrahimi-Fakhari D, Meyer S, Vogt T, et al. Dermatological manifestation of tuberous sclerosis complex (TSC). J Dtsch Dermatol Ges 2017;15:695–700. DOI: 10.1111/ddg.13264.
11. Ebrahimi-Fakhari D, Mann LL, Poryo M, et al. Incidence of tuberous sclerosis and age at first diagnosis: new data and emerging trends from a national, prospective surveillance study. Orphanet J Rare Dis 2018;13(1):117. DOI: 10.1186/s13023-018-0870-y.
12. Curatolo P, Jóźwiak S, Nabbout R. TSC Consensus Meeting for SEGA and Epilepsy Management. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol 2012;16(6):582–586. DOI: 10.1016/j.ejpn.2012.05.004.
________________________
© The Author(s). 2019 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.